|
Bios Dr. Fortenbach is an DISCLOSURES: The authors have no relevant relationships to disclose. |
A 45-year-old woman was referred to our tertiary care hospital for a second opinion on refractory central serous chorioretinopathy in the right eye for one year. She reported an initial decline in vision three years prior to presentation and was seen by a local retina specialist who started her on oral eplerenone 50 mg daily, which resolved her symptoms. About two years later, during a period of significant social stress, she experienced worsening vision. There was no improvement with additional eplerenone, and she didn’t tolerate acetazolamide 250 mg orally. At the time of presentation, she had continued blurry vision in her right eye for about one year without improvement. She was otherwise healthy and denied caffeine consumption, prior steroid use and history of cancer. Review of systems revealed a negative sleep study for obstructive sleep apnea.
Examination findings
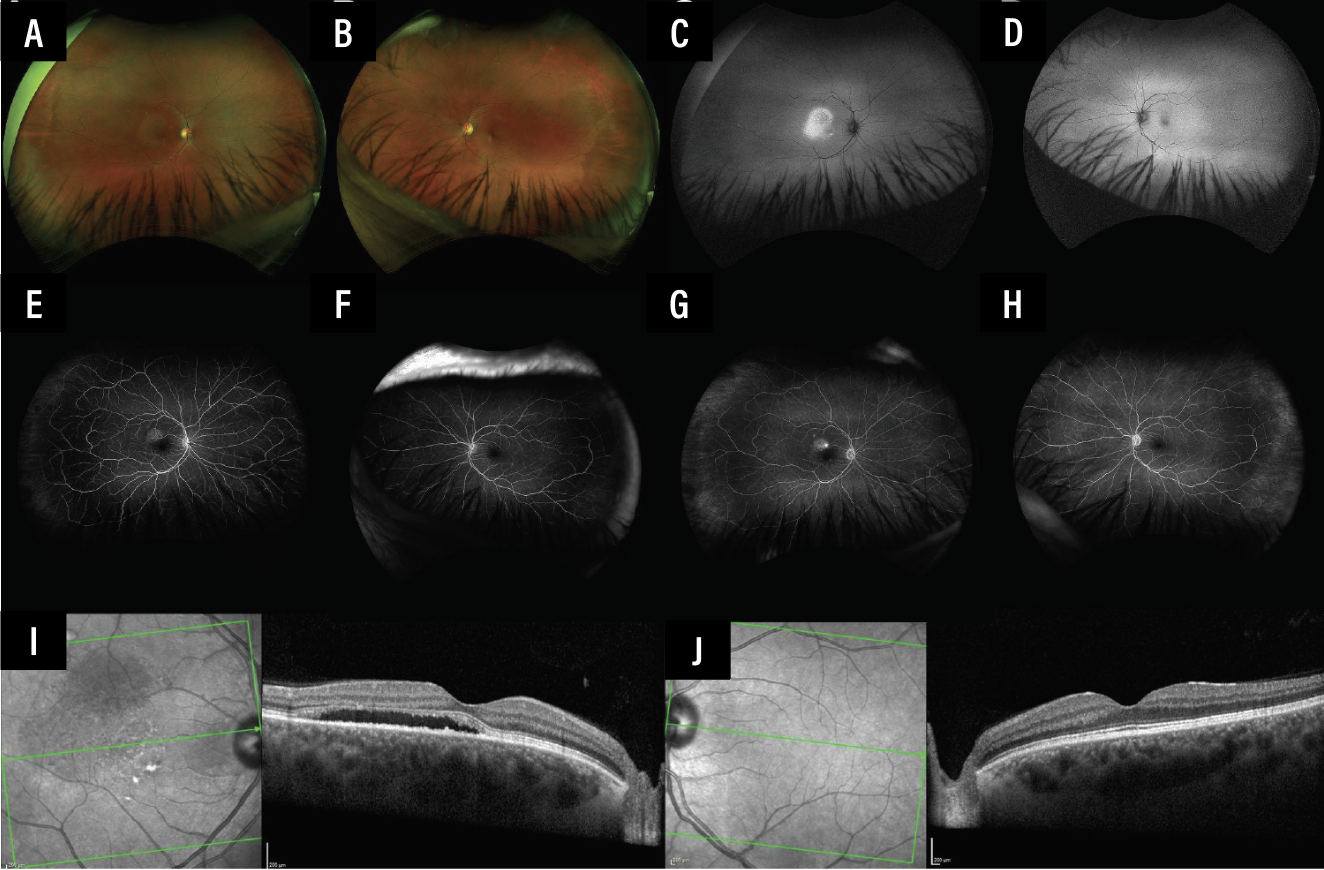
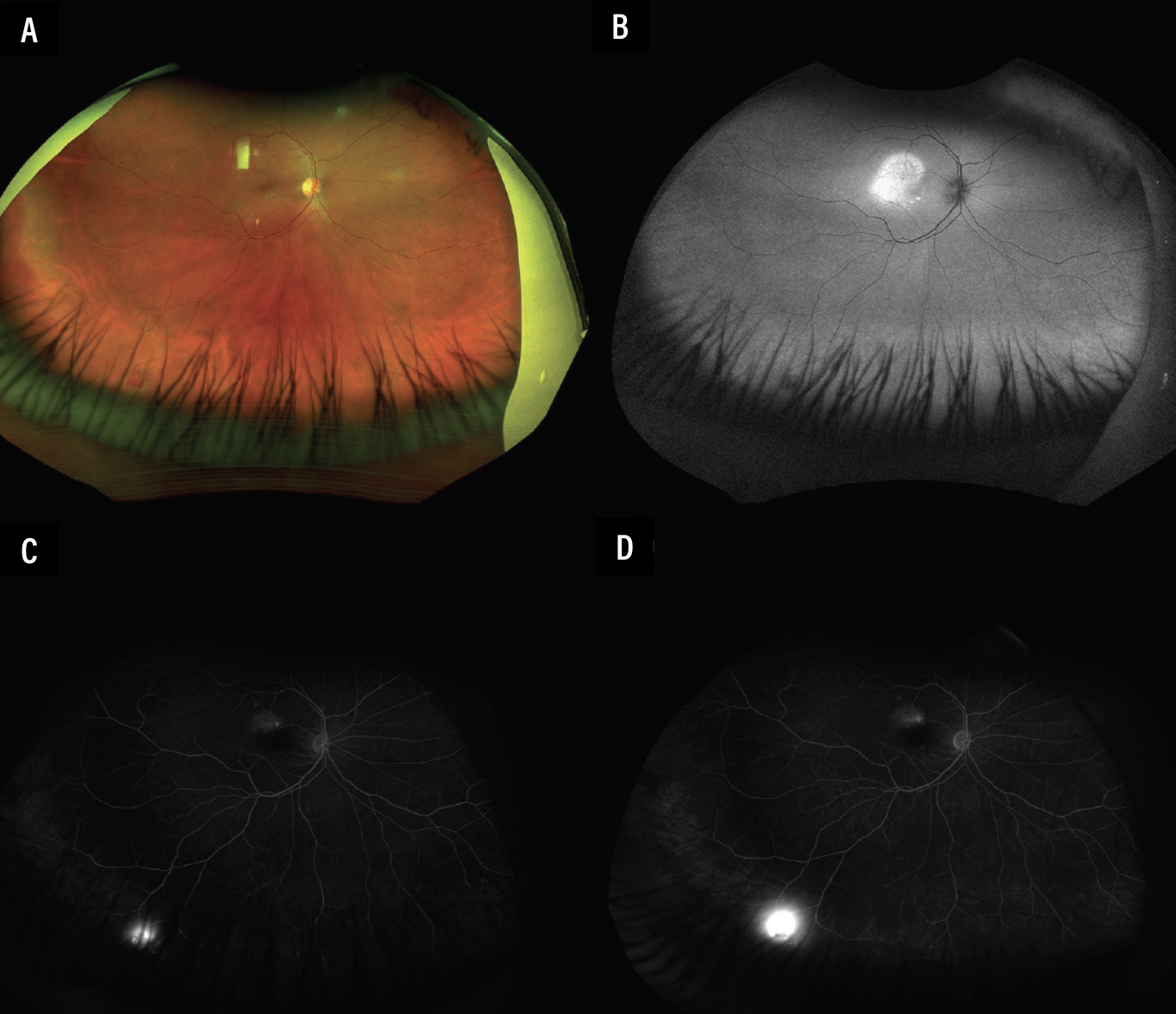
Her presenting visual acuity was 20/20 in both eyes with normal intraocular pressures and pupillary responses. Slit lamp biomicroscopy revealed an unremarkable anterior segment. Dilated fundus examination showed an unremarkable left eye but subretinal fluid in the macula and an orange vascular lesion in the inferotemporal periphery of the right eye. Pseudocolor fundus photography showed SRF in the right macula with a small one-disc diameter vascular lesion in the far inferotemporal peripheral retina (Figures 1A and 2A). There was also hyperautofluorescence corresponding to the region of SRF in the right macula (Figures 1C and 2B). The left eye was unremarkable (Figure 1D).
Workup
Fluorescein angiography showed early hyperfluorescence from the inferotemporal peripheral lesion, with marked leakage at later phases in the right eye (Figures 1E-G and 2C-D). FA of the left eye was unremarkable (Figures 1F and 1H). Spectral-domain optical coherence tomography of the right eye showed SRF without intraretinal fluid or retinal pigment epithelial detachments (Figure 1I). SD-OCT of the left eye was normal without SRF (Figure 1J).
 |
|
Figure 1. Multimodal imaging of the right and left eye. Pseudocolor fundus photography shows SRF OD (A) and an unremarkable left eye (B). There’s corresponding fundus hyperautofluorescence OD (C) and normal autofluorescence OS (D). FA shows early hyperfluorescence (E) and leakage (G) OD with normal fluorescence appearance OS in early (F) and late phases (H). Of note, in the late-phase fluorescein angiogram of the right eye (G), there is subtle but notable hyperfluorescence in the inferotemporal periphery. SD-OCT reveals SRF with a shaggy ellipsoid zone OD (I) and normal retinal laminations OS (J). |
Diagnosis and management
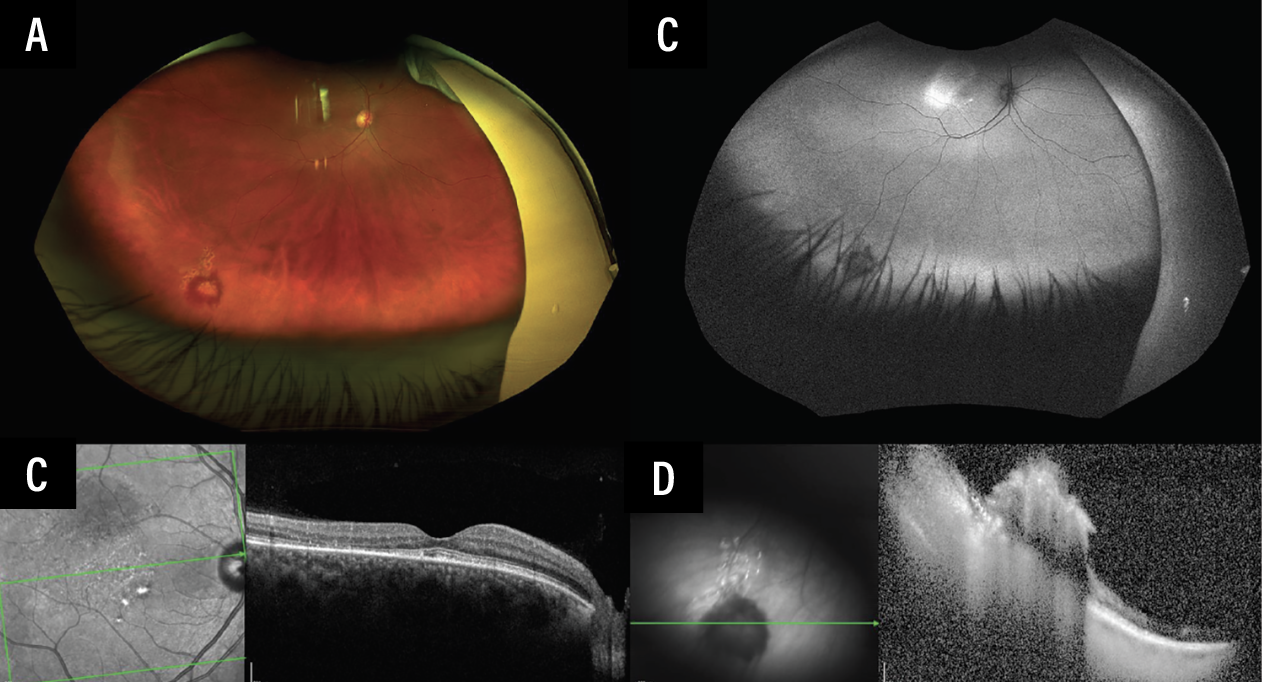
The prior diagnosis of central serous chorioretinopathy came into question, and a new unifying diagnosis was considered: chronic macular SRF associated with a peripheral leaking retinal vascular lesion. This hypothesis could be best tested by treating the vascular tumor. Grid laser photocoagulation targeting the tumor surface as well as the feeder arteriole was performed (Figures 3A-B). Eight weeks later, the patient’s visual acuity remained stable at 20/20, and her SRF had completely resolved (Figures 3C-D). Genetic testing for a VHL mutation was negative.
 |
|
Figure 2. Pseudocolor fundus photography of the right eye in downgaze. This eye shows a vascular lesion in the inferotemporal peripheral retina (A), which appears hypoautofluorescent (B). FA in early phases reveals early hyperfluorescence (C) with marked leakage in later phases (D). |
Discussion
This case demonstrates a leaking retinal vascular tumor masquerading as chronic, treatment-resistant central serous chorioretinopathy. While various retinal vascular tumors exist, including Coats’ disease and vasoproliferative tumors, this lesion is most consistent with a sporadic retinal capillary hemangioma. These are vascular hamartomas of the retina that manifest as reddish-orange vascular tumors and may be unifocal or multifocal, unilateral or bilateral and are characteristically associated with a markedly dilated feeding artery and draining vein. They may occur anywhere on the fundus, including the macula, equator, peripheral retina and rarely the optic disc. FA demonstrates rapid early hyperfluorescence and bright late leakage. These tumors may be asymptomatic or can cause vision loss from exudation, retinal tractional changes, vitreous hemorrhage or neovascular glaucoma.
 |
|
Figure 3. Status-post grid laser photocoagulation over and surrounding the retinal vascular lesion with its feeder arteriole in the right eye. Scanning laser ophthalmoscopy of the inferotemporal periphery OD shows laser over and around the retinal vascular lesion (A). The lesion is surrounded by hypoautofluorescent laser scars (B). SD-OCT through the right fovea shows resolution of SRF with loss of temporal outer retinal layers (C). SD-OCT through the hemangioma reveals findings consistent with an elevated retinal vascular lesion (D). |
These lesions may be sporadic or associated with Von Hippel-Lindau disease. Also known as familial cerebro-retinal angiomatosis, VHL is associated with an autosomal dominant mutation to the tumor suppressor gene VHL on chromosome 3p26-p25.1 Retinal lesions are often first visible at age 10 to 35 years, 25 years being the average, and are bilateral in 50 percent of cases with a third of patients having more than one lesion in the same eye.2 All patients with retinal capillary hemangioblastomas should undergo genetic testing for mutations to the VHL gene, as VHL is associated with other systemic tumors that can be fatal if not addressed. The most common causes of death from VHL disease are central nervous system hemangioblastomas (cerebellum, medulla, pons, spinal cord) and renal cell carcinoma. Other associated tumors include pheochromocytoma, endolymphatic sac (inner ear) tumors, cardiac rhabdomyomas, renal, pancreatic cysts, liver cysts and bilateral papillary cystadenomas of the epididymis or the broad ligament of the uterus. All carriers of the VHL mutation should undergo screening for tumors of the kidney, pancreas, inner ear, retina and central nervous system.
The management of retinal capillary hemangioblastomas depends on tumor size, location and related features of SRF and exudation.3,4 Small-sized tumors (<3 mm in diameter) can be treated with laser photocoagulation that surrounds the tumor and closes the feeding artery. Medium-sized tumors (3 to 6 mm in diameter) can be treated with verteporfin photodynamic therapy if posterior to the equator or with cryotherapy if anterior to the equator. Large-size tumors (>6 mm in diameter) may require radiotherapy with plaque brachytherapy, external beam radiotherapy, proton beam therapy or vitrectomy with internal resection and silicone tamponade. Intravitreal anti-vascular endothelial growth factor can be attempted to reduce subretinal and intraretinal fluid, but often shows little effect on the tumor. Juxtapapillary tumors present a treatment dilemma due to their proximity to the papillomacular bundle, but photodynamic therapy can be attempted in these cases.5
More recently, the oral hypoxia-inducible factor 2α inhibitor, belzutifan, which received FDA approval for the treatment of central nervous system and renal hemangioblastomas associated with VHL disease, has been investigated for the treatment of retinal capillary hemangioblastoma. The LITESPARK-004 study has demonstrated the efficacy of oral belzutifan in the regression of retinal hemangioblastomas in patients with VHL disease, as have several case reports.6-7
Bottom Line
This case highlights the importance of avoiding an anchoring bias towards the initial diagnosis when evaluating a new patient. It’s essential to do a complete and thorough workup and examination for all first-time patients, including a detailed peripheral retinal examination. While the leaking retinal capillary hemangioma detected in this case was subtle and unexpected, the ability to treat it ultimately relieved the patient’s chronic SRF. RS
References
1. Chittiboina P, Lonser RR. Von Hippel-Lindau disease. Handb Clin Neurol. 2015;132:139-156.
2. van Leeuwaarde RS, Ahmad S, van Nesselrooij B, Zandee W, Giles RH. Von Hippel-Lindau syndrome. In: GeneReviews. University of Washington, Seattle; 1993.
3. Singh AD, Nouri M, Shields CL, Shields JA, Perez N. Treatment of retinal capillary hemangioma. Ophthalmology. 2002;109:10:1799-1806.
4. Won YK, Lee MW, Shin YI, Kim JY, Jo YJ. Clinical results of various treatments for retinal capillary hemangioma. Korean J Ophthalmol. 2020;34:2:133-142.
5. Mohamad NM, Shokri SS, Sukaimy FS, Wan Muda WN, Ahmad Tajudin LS. Therapeutic dilemma of a juxtapapillary retinal capillary hemangioma. Cureus. 2024;16:12:e76598.
6. Wiley HE, Srinivasan R, Maranchie JK, et al. Oral hypoxia-inducible factor 2α inhibitor belzutifan in ocular von Hippel-Lindau disease: Subgroup analysis of the single-arm phase 2 LITESPARK-004 study. Ophthalmology. 2024;131:11:1324-1332.
7. Mustafi D, Huang J, Ting MA, Waligorski N, Stacey AW, Huang LC. Successful treatment response of a juxtapapillary retinal capillary hemangioblastoma due to von Hippel-Lindau syndrome with belzutifan in a pediatric patient. Retina. 2024;44:5:e31-e33.